- History Home
- People, Leadership & Service
- A Legacy of Excellence
- History & Impact
- Meetings Through the Years
- Resources
Memoir - Jack D. DunitzMemoir | Publications | Curriculum Vitae | Videos | Slides | Articles |Obituary
La Primavera: An Autobiographical Essay Jack D. Dunitz Alas, that Spring should vanish with the Rose Edward Fitzgerald, 1809-1883
Jack Dunitz receiving the 1991 ACA Buerger Award from ACA President Judith Flippen-Anderson.
It is a delicate matter to write about oneself, about one's plain own achievements. It is difficult to attain and maintain the necessary degree of objectivity that keeps a balance between under- and overestimation of one's abilities, between undue modesty and self-satisfied complacency. Even if one tries to be objective about the past, memory is fallible. The lens through which it views the past is selective and liable to omissions, extensions and plain mistakes. From a distance we look backwards with a false and distorting perspective, which may sometimes make things better than they actually were, and sometimes worse. Yet, fully aware of all these difficulties and dangers, I feel a certain responsibility to try to account for my actions. In this past terrible century, where so many people have undergone appalling suffering, I have enjoyed the privilege of a long and happy life. In a century where so many people were destined to be murdered or murderers, I was fortunate in escaping both these destinies. My life has been free from tragedy for me and my family, I have never suffered the pangs of real hunger, have lived for the most part in comfort and plenty, have enjoyed good health, and have not been condemned to a job I hated or even one in which I was bored. In fact, I have had the enormous good fortune to spend my time more or less as I liked to spend it — in inventing and solving scientific problems, mainly in structural chemistry, problems that to the best of my knowledge have had no practical relevance, neither for good nor for evil. The last half-century has probably been the only time in history when such a thing was possible, when a person of modest abilities could enjoy a comfortable existence doing whatever seemed interesting. I feel I have been tremendously fortunate.
After some self-questioning, I decided to agree to write something about my history but to keep to the first, itinerant stage of my scientific life — Glasgow, Oxford, Pasadena, Oxford, Pasadena, Bethesda Maryland, London — the springtime of my life, the period before I settled down in Zurich. That early part is divided into sub-periods, each lasting a couple of years, which stand out separately so that each seems have an own individuality, whereas the subsequent three decades of summer and two of autumn fall merge into a sort of continuum in which it is more difficult to distinguish any underlying pattern, except that in 1990 my official position as professor at the ETH Zurich ended. In fact, during the Zurich period, it is the absences from Zurich that stand out — visiting appointments in different parts of the world, travels to international meetings — the events that break the uniformity of the general background in which, of course, most of my work was done. Another reason for concentrating on my early years is that I feel I can be more objective towards that young scientist stumbling over his first steps in science than towards my more recent self. Indeed, when I see myself in photographs from that period (and earlier), I experience very little connection between that person in front of the camera and my present day self. It is as if I were looking at a portrait of someone else, someone I used to know fairly well, but whom I haven't seen for a long time. When I have told friends about my lack of identification with my former self, I have been warned that this kind of self-estrangement is a symptom of possibly severe psychoanalytical disturbance. I considered the possibility of writing this memoir about myself in the third person: Jack did this and then Jack did that, but when I wrote some sentences in that style it seemed unnatural.
I do not remember ever having made a conscious choice or decision to follow a career in science. It just happened, as in a dream. In a dream you don't do things, things happen to you. Near the beginning, during the early 1940's, I was a student of chemistry at Glasgow University, ignorant not only of chemistry but of almost everything else, an innocent in every way one can imagine. I did not choose chemistry. My mother would have liked me to study medicine, my headmaster tried to push me into the study of classics. I did not much fancy either of these possibilities. The science teacher at my school, John McLennan, chose chemistry for me by making it interesting. Likewise, I did not choose crystallography. Crystallography chose me. As a frustrated mathematician, my interests were mainly in physical chemistry. After a somewhat compressed wartime three-year crash course in chemistry, most of the Phys. Chem. Students were funneled off for work about which they were not allowed to talk (now we know it was radar research), but in 1943 John Monteath Robertson returned to Glasgow as newly appointed Gardiner Professor and needed a few doctoral students to carry out work in X-ray crystallography and molecular structure studies. At that time and place, there was no question of a student choosing a research supervisor or a line of research. It was still wartime. "You, you and you will report for duty at such and such a locality, you, you and you will stay on here and work for Robertson" and so it was that I came to chemical crystallography. One of the others directed to work for Robertson was John White. Later, as assistant professor at Princeton, he started a program of research on the crystal structure elucidation of vitamin B12, quite independently of Dorothy Hodgkin's work on the problem in Oxford. When they learned of each other's work, they combined their results. The first publications on the work were joint papers, from both groups. It seems ironic that for their respective contributions Dorothy was eventually awarded the Nobel Prize while John was not given tenure at Princeton. Later, he became a professor at Fordham University, New York. Another student sent to work with Robertson was Ian Dawson, who later joined the faculty at Glasgow, where he developed electron microscopy and was the first to observed growth spiral steps in crystals of long-chain hydrocarbons. After a few months we were joined by Alexander (Sandy) Mathieson, who later emigrated to Australia and built up a strong chemical crystallography group at the CSIRO in Melbourne. We had a wonderful time together. As Robertson was much away on official duties, and as there was no formal post-graduate course of study, we taught one another what we had taught ourselves about the theory and practice of crystal structure analysis.
And so it went on. In the autumn of 1946, chance took me to work with Dorothy Hodgkin at the Chemical Crystallography Laboratory at Oxford. In early 1948, Linus Pauling came to Oxford as Eastman Visiting Professor. Through the happy intervention of his younger colleague Verner Schomaker, who visited our laboratory on an early summer afternoon when Dorothy was absent and talked to me instead, I was invited to be a research fellow at Caltech (1948-1951). Then back to Oxford (1951-1953), then back to Pasadena for a further year, then to the U.S. National Institutes of Health in Bethesda, Md. (1954-1955), and then in 1956 to the Royal Institution in London, where Sir Lawrence Bragg, recently appointed as Director after his retirement from his professorship at the Cavendish Laboratory in Cambridge, was recruiting a group of young crystallographers. After only a year and a half in London, Ilanded in 1957 at the Organic Chemistry Laboratory of the ETH in Zurich, to join an illustrious group of natural philosophers there, my friends and colleagues for the last fifty years and more, during which we have argued and discussed and learned together about chemistry and molecular structure and about everything else under the sun.
It seems like an erratic somnambulistic path. Among the things that were happening to me, did I make one single decision? Yes! In 1953 I decided to marry Barbara Steuer — or was that also decided for me? Our years together have made my life richer in countless ways. We have been blessed with two wonderful daughters, six wonderful grandchildren and one wonderful great granddaughter (so far, as of October 2012). I am also fortunate in having been taught over the years by a series of students, postdocs, and assistants, from whom I have learned nearly everything I know. It has been my privilege to share in the development of our science during the last half-century and more. I do not regret a minute of it. If I had another life I would be happy to live it along much the same lines as I have lived this one.
 Barbara S. Dunitz and Jack D. Dunitz, 1985. (Courtesy of Carolyn P. Brock)
FIRST STEPS IN SCIENCE, 1943-1946
For my doctoral work at Glasgow my first task was that of determining the crystal structures of acetylenedicarboxylic acid. The reason for this had something to do with the anomalous isotope effect observed some years earlier in crystals of oxalic acid dihydrate,[1] where deuterium substitution led to a slight expansion of the structure in the direction of the main hydrogen bonds instead of the usual contraction. With today's methods the project could be completed in a morning. It took me about three years. In that time I was able to show that the two acid dihydrates had very similar crystal structures and that diacetylenedicarboxylic acid dihydrate, the next member of the series, had a similar but interestingly different structure. Whereas the first two crystals contained centrosymmetric, coplanar molecules, diacetylenedicarboxylic acid molecules were situated on dyad axes and the two carboxylic acid groups were twisted with respect to each other. Nevertheless, the hydrogen bonding network remained very similar in all three structures.[2] We never did get so far as to study the anomalous isotope effect in these crystals. A few years later, a theoretical explanation was given by Nordman and Lipscomb[3], so it ceased to be anomalous, although I am not convinced that the explanation offered is correct. And fifty years later, I rewrote my first three papers in more modern chemical parlance; the new title was "A Supramolecular Three-Dimensional Hydrogen-Bonded Network with Potential Application in Crystal Engineering Paradigms".[4] Some readers thought it was frivolous but it was meant to be taken seriously.
OXFORD (1946-1948)
Although J. Monteath Robertson had invented the method of isomorphous replacement, we never used it during my years at Glasgow. For the sort of problem I was working on, there was no opportunity to apply the heavy atom method — there were no heavy atoms in the compounds studied — nor to make use of the Patterson function. What we did learn was how to look at an X-ray diffraction diagram and see the main features of the molecular transform, an ability that proved to be very useful, both then and later. One day during my Glasgow work, I read Dorothy Crowfoot's paper on the structure of cholesteryl iodide.[5] This was one of the first three-dimensional analyses of what was then regarded as a complex molecule — the penicillin work was not yet published. I wrote to Dorothy Hodgkin (neé Crowfoot) to ask if she would accept me as a post-doctoral research worker, and she agreed to do so. I had no money, but the Carnegie Trust for the Universities of Scotland, which had provided financial support to me as a research student in Glasgow, generously decided to continue and even to augment my stipend.
After my arrival in Oxford in late 1946, my first project was to try to determine the detailed crystal structure of a calciferol derivative. From earlier comparisons of unit cell dimensions of sterols and related compounds, it had been tentatively concluded that the calciferol molecule, in spite of its broken B ring, had more or less the same shape as a normal sterol, such as cholesterol.[6] I was sure this must be incorrect. As Dorothy herself was doubtful about the matter, we decided to try to obtain a suitable heavy atom derivative. Fortunately, we were able to persuade John (Kappa) Cornforth and his wife Rita to help us, but it took several months before they could come up with a suitable crystalline derivative, the 4-iodo-5-nitrobenzoate of calciferol. From a two-dimensional projection down the short unit cell axis,[7] it was clear that the molecule was indeed s-anti about the central bond of the broken B ring and not s-syn, as had been assumed in the early work. With more than 40 atoms (not counting hydrogens) in the molecule, this was at that time the most complex structure to have been determined at atomic resolution — a record that did not endure very long. I was able to do the calculations required for the two-dimensional projection by hand, with the help of Beevers-Lipson strips[8] — does anyone still remember what they were? — but the completion of the full three-dimensional analysis had to wait many years until appropriate computing facilities became available.[9]
During the waiting time for suitable crystals, we had a visit from a member of the Medical Research Council staff. It had been found that the drug stilbamidine, a 4,4'-diamidostilbene derivative, undergoes photochemical change to a more toxic substance when its solutions are exposed to light. The toxic product could be converted to a hydrocarbon, C28H24, melting temperature 163°C, identical to the product obtained many years earlier by irradiation of stilbene itself[10] and identified as 1,2,3,4-tetraphenylcyclobutane, together with a small amount of an isomer, melting temperature 149°C. The question of interest to the Medical Research Council was: Could we tell which stereoisomer was which? From the unit-cell dimensions and space group information, the higher melting compound was soon identified as the centrosymmetric isomer. As I had nothing better to do at the time, I practiced my skill in trial-and-error analysis by determining the crystal structure in projection down the short (5.77 Å) monoclinic axis[11] and then also tested my endurance by calculating lines and sections of the three-dimensional electron density distribution, based on visual estimates of all photographically recorded reflections within the CuKα sphere of reciprocal space.[12] From today's perspective, when the measurements could be made in a few hours and the calculations in a few seconds, it is hard to imagine how much drudgery was involved in such an exploit in those days, working with paper, pencil and Beevers-Lipson strips. Why on earth did I take it on, and why did I persist? No one was pushing me. Perhaps I merely wanted to show that I could do it.
Fame is the spur that the clear spirit doth raise (That last infirmity of noble mind) To scorn delights and live laborious days.
My tetraphenylcyclobutane work did not bring me fame but it did bring me to Caltech. Indirectly. From my results, the bond distances in the cyclobutane ring appeared to be longer than the standard carbon-carbon single-bond distance of 1.54 Å, while those in the phenyl groups were normal for benzene rings. However, according to the recently developed "bent bond" model[13], bonds in small carbocyclic rings were expected to be slightly shorter than 1.54 Å, as had been found, indeed, for cyclopropane and spiropentane from gas-phase electron diffraction.[14] Were the long bonds found in tetraphenylcyclobutane an intrinsic property of the cyclobutane ring? Or were they in some way connected with the presence of the four phenyl substituents? Or were they merely attributable to experimental error? When I discussed this problem with Verner Schomaker during his visit to Oxford in the early summer of 1948, we decided that the problem called for a gas-phase electron diffraction study of cyclobutane itself. I was interested in learning this technique, and, through Schomaker's intervention, Pauling offered me a research fellowship to come to Caltech.
At Oxford, I became friends with Gerhard Schmidt, who, although a few years older than I, was still working for his D. Phil. Schmidt, a Jewish émigré from Germany, had lost valuable years, as he been arrested in 1940 as an "enemy alien", sent to the internment camp in the Isle of Man and then deported to Australia. Eventually, as a result of intervention by Sir Robert Robinson, Professor of Organic Chemistry at Oxford University, he was able to resume his studies. Schmidt had learned his organic chemistry from Robinson and did his best to improve and extend my more limited and old-fashioned views on the subject. We often worked into the night. Gerhard went on to found that marvelous school of structural chemistry at the Weizmann Institute. He told me the following story, which deserves to be secured for posterity. In one of his Oxford lectures, Robinson had drawn on the blackboard an organic formula with a pentavalent carbon atom. After some hesitation as to whether he should interrupt his famous and reputedly irascible teacher, Gerhard tentatively raised his hand. Robinson stopped. "Well," he asked, with a disapproving stare. "Excuse me, Professor, but in the alkaloid structure on the left of the board you have drawn a pentavalent carbon atom". Robinson glared at Gerhard, turned to the board, erased the superfluous bond, glared again at Gerhard, and continued with the lecture. Twenty years later, during a visit to the Weizmann Institute, Sir Robert again wrote an incorrect formula in the course of a lecture. By this time, Gerhard was himself a well-known scientist and had enough self-confidence to immediately draw the lecturer's attention to his error. Robinson glared at Gerhard, turned to the board, glared back at Gerhard, and exclaimed: "You again, Schmidt!"
PASADENA (1948-51, 1952-1953)
Those of us who had the privilege of working at Caltech during those years refer to that period as the Golden Age. Apart from the great Linus Pauling, there was a U.S.A. select team in structural chemistry: Verner Schomaker, Eddie Hughes, Robert Corey, Jerry Donohue, Kenneth Trueblood, Gene Carpenter, Ken Hedberg, David Shoemaker, Alex Rich, Dick Marsh, besides a stream of post-docs from outside the U.S.A.; Gunnar Bergmann from Sweden, Otto Bastiansen from Norway, Edgar Heilbronner from Switzerland, Massimo Simonetta from Italy, You-chi Tang from China, all in the first period, John Rollett, David Davies, Leslie Orgel, all from England, Giovanni Giacometti from Italy in the second. Among the graduate students were Walter Hamilton, Doyle Britton, Martin Karplus, Jim Ibers, Hans Freeman, Berni Alder and Joe Kraut. All of these became future leaders in structural chemistry and cognate areas of research. James A. Ibers and Jack D. Dunitz in Zurich, 1989. (Courtesy of Carolyn P. Brock)
Right from the start it was obvious to me that my Caltech colleagues had a much better background in theoretical crystallography, diffraction physics and mathematics than I had; even the graduate students seemed to know more about these matters than I had learned in my five years of practice at Glasgow and Oxford. I saw that one of my first priorities would be to establish a more solid base in these subjects for myself. Fortunately for me, my new colleagues were generous in sharing their knowledge.
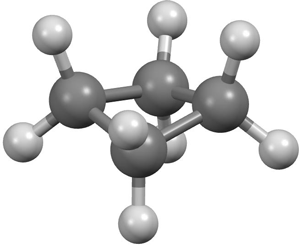
The cyclobutane structure was to be my major research task. Verner arranged with colleagues in Berkeley for a small cylinder of the material to be delivered to us. Meanwhile, he and Kenneth Hedberg introduced me to the theoretical and experimental aspects of gas-phase electron diffraction. I shall not dwell here on these, except to mention the curious fact that our visual estimation of the scattered intensity was actually based on an optical illusion. The diffraction patterns, recorded on photographic plates, appeared to the eye to consist of a set of concentric rings of gradually decreasing intensity. According to photometric measurement, however, the scattered intensity showed a monotonic decrease from the film centre outward, with no outspoken maxima or minima. The eye automatically compensates for uniform intensity fall-off and interprets differences in the rate of change between neighboring regions of the film as local maxima and minima. The first step for the novice was to learn to translate the visual experience of rings of high and low intensity into a physically interpretable diffraction pattern. This was achieved by looking at photographic diffraction patterns from diatomic molecules (chlorine, oxygen) until one "saw" the intensity variation predicted by theory: a set of equidistant rings corresponding to the single frequency produced by the single interatomic distance. After a few months I was admitted into the small, select circle of people whose opinion about features on electron diffraction photographs was deemed to matter, and, by the end of the year, as a prelude to the planned cyclobutane analysis, I was immersed in the interpretation of photographs obtained previously for cyclopropene. Then I helped Hedberg to redetermine the structural parameters of the triatomic molecules ClO2 and C12O.[15] It is remarkable how well our results derived by such primitive methods compare with those from analyses made many years later with more up-to-date equipment. Model of buckled cyclobutane. (Courtesy of Jeff Deschamps)
Our gas-phase electron-diffraction study of cyclobutane[16] confirmed my supposition that the C—C bonds were long. Moreover, contrary to what had been generally assumed until then, the four-membered ring was not a planar square but was buckled (D2drather than D4h symmetry). The reason for the striking difference between the carbon-carbon bond distances in cyclopropane (1.51 Å) and cyclobutane (1.57 Å) is that in the former there are no non-bonded 1,3-interactions, whereas the four-membered ring shows the strongest possible interactions of this type, which are, of course, strongly repulsive. It was a great experience to work with Verner Schomaker, to argue with him, and, more than anything, to share with him the writing of a scientific paper. Our cyclobutane paper took ages to write, but, as compensation, after almost 50 years, I am still pleased with the result. Among other things, that publication contained what must have been one of the earliest force-field calculations, and also a carefully qualified sentence defining what we meant by the term "bent bond": "It appears that this argument might be expressed in terms of the significant existence of a bond line, to be distinguished from the internuclear (straight) line, which more or less follows a line of maximum density of the bonding electron distribution, and which, in the bent bond, tends to retain a fixed length, thereby possibly causing the internuclear distance to be shortened in spite of the resulting increased internuclear repulsion." More than thirty years later, when bent bonds had become fashionable and had showed up in Bader's theory of chemical bonding, this definition received a seal of approval when it was reproduced in one of Bader's papers[17]— although without the commas over which we had deliberated so long. Not many sentences in the scientific literature are deemed to be worth repeating after thirty years.
Apart from my work in the chemistry laboratories, I spent a fair amount of time with members of Max Delbrück's phage group, especially with Gunther Stent, Carleton Gajdusek, Jean Weigle and Elie Wollman. A feature of this group was the active social life enjoyed by its members. In addition to frequent parties at the Delbrück home, there were regular expeditions to destinations in the nearby California deserts and mountains. After driving along perilous dirt roads until a suitable campsite was reached, we cooked our meals on an open fire, drank copious amounts of cheap, sold-by-the-gallon wine, and slept under the brilliant stars. Topics of discussion around the campfire were not restricted to scientific ones; they included such matters as the superiority of Mozart over Wagner, the authorship of Shakespeare's plays, mathematical puzzles (such as the generalized cake problem), and many, many others.
Among these was the fundamental problem: How do living organisms manage to make copies of themselves? How is the organizational information stored and transmitted down the generations? Of course, we did not know the answer. It must have been early in 1949 when Delbrück told me that in his opinion my work in structural chemistry was a waste of time. He proposed that I should abandon it and join his research group instead; the switch to phage work would provide an entrance into the world of biological research and enable me to use such talents as I possessed to better purpose. I was dismayed that Delbrück thought so little of my work in chemistry but at the same time flattered that he saw me as having potential for creative research in his own area, for he was known to have extremely high intellectual standards. After a couple of weeks I decided not to accept his offer. I felt I had just reached the stage in my own development where I was reasonably competent in at least one area of research; I knew where the good problems lay and I knew I could solve them as well as anyone else. Having just reached that stage, I was unwilling to start again as a novice in a field where I was essentially ignorant. I did not want to be an eternal student, at least not only an eternal student.
I recalled this decision nearly twenty years later, when Delbrück sent me a copy of the Festschrift "Phage and the Origins of Molecular Biology" dedicated to him on his 60th birthday.[18] By that time the revolution in molecular biology had occurred, and Max was being celebrated as one of its founders. I wrote to express my appreciation that he had once thought of me as a potential collaborator and reminded him of his offer and of my reasons for declining it. I told him I had no regrets; I felt I had made the right choice and I still do so.
I have never regarded Delbrück as one of the fathers of molecular biology. In fact, in those days it was my impression that he was rather hostile to the ideas behind molecular biology. As I recall, he sat beside me at the lecture where Pauling first publicly announced his stable hydrogen-bonded model structures for polypeptide chains. Pauling had a feeling for drama. On the table in front of him stood bulky columnar objects shrouded in cloth, which naturally excited the curiosity of those in the packed auditorium. Only after describing in detail the structural principles behind the models did he turn to the table and unveil the molecular models with a characteristic theatrical gesture. There were the two structures, the three-residue and the five-residue spirals, later dubbed the α- and g-helices! I was immediately converted, a believer right from the start. In contrast, Max made no secret of his skepticism and especially his disapproval of Pauling's manner of presentation, and asked if I thought there was anything of value in these models. I believe I may have disappointed him when I told him that in my opinion the models were based on sound structural principles and were very likely to represent important building blocks of actual proteins.
While my own work at Caltech had nothing to do with protein structure, Pauling used to talk to me occasionally about his models and what one could learn from them. In his lecture, he had talked about spirals. In conversation a few days later, I told him that for me the word "spiral" referred to a curve in a plane. As his polypeptide coils were three-dimensional figures, I suggested they were better described as "helices". Pauling's erudition did not stop at the natural sciences. He answered, quite correctly, that the words "spiral" and "helix" are practically synonymous and can be used almost interchangeably, but he thanked me for my suggestion because he preferred "helix" and declared that he would always use it henceforth. Perhaps he felt that by calling his structure a helix there would be less risk of confusion with the various other models that had been proposed earlier. In their 1950 short preliminary communication, Pauling and Corey wrote exclusively about spirals[19], but in the series of papers published the following year[20]the spiral had already given way to the helix. There was no going back. A few years later we had the DNA double helix, not the DNA double spiral. The formulation of the α-helix was the first and is still one of the greatest triumphs of speculative model building in molecular biology, and I am pleased that I helped to give it its name.
The structural chemistry group at Caltech was among the first to use punched-card methods to ease the computational problems of crystal structure analysis. We could complete a three-dimensional structure factor calculation or Fourier synthesis for a small-to-medium organic structure overnight instead of over a period of weeks. During my first stay at Caltech I made a simple program for two-dimensional least-squares analysis in space group P21/a, and during my second stay I helped John Rollett to write a general least-squares program, including refinement of "thermal" as well as positional parameters, for the impressive sounding Electrodata Datatron, an early electronic computer, which we were allowed to use after normal office hours, that is, during the night, from evening until early morning. My contribution to this was quite modest. John wrote the program code during the day, and I called the numbers out to him in the evening as he typed them onto a tape. Together we worked on the crystal structure of dibenzyl phosphoric acid. Our objective was to determine the geometry of the phosphate di-ester group for comparison with the dimensions assumed in the Crick- Watson DNA model.[21] Nowadays, we would describe the C-O-P-O-C conformation in terms of the two C-O-P-O torsion angles, but at that time the utility of torsion angles as conformational descriptors had not yet come into general usage. At any rate, torsion angles are not mentioned in our paper.[22] Instead we discussed the dihedral angle between the two POC planes. We wrote then that the value of this dihedral angle "is 88° in dibenzyl phosphoric acid but only 63° in the Crick-Watson model" and proceeded to give an explanation of this difference "in terms of van der Waals repulsion between the phosphate group and the deoxyribose ring".
It is embarrassing to admit that we must have made a mistake in calculating the value of the dihedral angle in our structure. The correct value, based on our published atomic coordinates, is 65°, practically the same as the assumed Crick-Watson value. After so many years, I cannot remember how we calculated the value of this dihedral angle, nor can I find any record of how it was done. Presumably, one of us made the calculation and the other checked it, so we are both to blame. But before anyone censures us for having made such a grievous mistake, she should set herself the task of evaluating the dihedral angle in question with paper and pencil, without even the aid of a hand calculator, from the atomic coordinates and unit cell dimensions listed in our paper. As I recall, it was only years later in Zurich when we began to work on medium ring compounds that we began to use torsion angles systematically to describe the ring conformations and developed simple algorithms for calculating dihedral angles.
During my first stay at Caltech I was best known there not for any scientific accomplishments but for my collaboration with Ted Harrold, an English postdoc in George Beadle's research group, in writing and producing cabaret-style entertainments. Ted was an accomplished pianist who could improvise for hours in almost any style from Bach to Honkytonk, and we soon found a mutual fascination with the radio commercials that were so characteristic of popular culture in America at that time and a shared amusement in inventing parodies of them. Mostly we set new words to well-known tunes, but Ted also set some of our new words to music of his own. We thought the results were funny, and perhaps they were. Somehow or other, Beadle got to hear of this, and we were ordered, or let us say strongly encouraged, to organize an entertainment for the 1948 Departmental Christmas Party. Within a few weeks, we assembled enough material for a forty-minute cabaret and persuaded a few other people to join us in the songs and choruses. We asked our audience to imagine that radio commercials had been taken over for an evening by scientific and academic institutions eager to advertise their wares. The show included commercials from Harvard, Yale, Chicago, Caltech, the Sorbonne and even Oxford and Cambridge, as well as from manufacturers of scientific equipment. It was a great success and parts of it were later recorded. Ted and I produced shows for the two following Christmas Parties, in 1949 and 1950. These were probably not as successful as the first, although the 1950 production, a much-debased parody of Marlowe's Dr. Faustus, had some good lines in it.
OXFORD (1951-1953)
In the summer of 1950 I spent several weeks in Europe. At Oxford I met the young David Sayre, who told me about his approach to direct methods, which made a deep impression on me. When I returned to Oxford a year later, the calciferol analysis was at a standstill because of the false symmetry of the 3D-Fourier map that is introduced by the phasing from the heavy atom positions alone. I had the idea that it might be possible to break this false symmetry by extending Sayre's method to deal with non-centrosymmetric structures where the Fourier coefficients were associated with phase angles and not merely with signs, plus or minus. For this purpose I developed an equation that was essentially the same as the famous tangent formula developed by Karle and Hauptman. The first step was to make a list of "triples" of strong reflections with indices H, K, H-Kthat formed, together with the origin, parallelograms in reciprocal space. My plan was to do this graphically, by plotting in ink on transparent sheets the various layers of reflections, moving the origin to each reflection in turn and looking for coincidences. I sat upstairs in a kind of balcony in the large room in University Museum that housed Dorothy Hodgkin's research group, the same room in which Thomas Henry Huxley had publicly rebuked Bishop Wilberforce in a debate about evolution some 80 years earlier. There, day after day, I overlaid transparent sheet on transparent sheet, looking for parallelograms in three-dimensional reciprocal space. After about two months I took stock; I had completed about 10% of the preliminary, trivial, more or less automatic step in my new method. I estimated I would need to invest about two years of boring, repetitive work before reaching the stage where the method could be tested. I gave up. Ten years later we wrote a computer program that could do the job in a couple of seconds. But by that time the method had been shown to work[23] and the calciferol problem had in any case been solved by other means.8
I was in the habit of spending a few hours every week in the Bodleian Science Library reading the latest scientific literature. This was not as voluminous as it is today. One could then read, or at least scan, all the important chemical journals and still have time to do some work on one's own. One afternoon in late spring of 1952 I came across an astonishing proposal from a group of Harvard chemists for the structure of the recently obtained compound, C10H10Fe, an orange colored solid, volatile, insoluble in water but readily soluble in organic solvents.[24] The proposed structure consisted of two parallel cyclopentadienyl rings with the iron atom sandwiched between them.[25] The only physico-chemical evidence offered for this unprecedented structure was the infrared absorption spectrum, which contained, in the 3-4 μ region, a single, sharp band at 3.25 m, indicating the presence of only one type of C—H bond. It may be difficult today to appreciate just how surprising, unorthodox, even revolutionary, this structure was at the time. At any rate, my first reaction was one of extreme skepticism. On my way out of the Library I met my friend Leslie Orgel, at that time holder of a Research Fellowship at Magdalen College, and asked if he had seen the remarkable structure proposed in the latest JACS number. We retrieved the journal and re-read the article together. He was as skeptical as I was. When we learned that the compound was relatively easy to prepare in crystalline form, we decided to make it and determine the crystal and molecular structure. Or rather, since neither of us had access to facilities in a synthetic laboratory, we decided to try to persuade a friendly organic chemist to carry out the relatively straightforward synthesis. In the nearby Dyson Perrins Laboratory we were fortunate to meet Hugh Cardwell, who agreed that the problem was worthwhile and offered to synthesize a few grams of this unusual and provocative compound. Within a week or two he fulfilled his promise.
According to the record in my laboratory notebook, I made optical measurements on crystals of the new compound on June 9th, 1952 and began to make preliminary X-ray photographs the following day. I soon found that the crystals slowly sublimed in the atmosphere at room temperature and had to be sealed into glass capillary tubes. From the space group alone it was evident that the molecule must sit at a crystallographic centre of symmetry. By the end of the following week, I had made enough intensity measurements to produce two electron-density projections down mutually perpendicular directions. This was possible because, fortunately for me, there was a slack period in the laboratory so that not only one but two X-ray Weissenberg cameras were free for my use. Of course, to get all this done on my own, I had to work long hours, during the evenings and over the weekend too. As the structure began to emerge from the electron-density maps, calculated with Beevers-Lipson strips with the aid of an adding machine, I was becoming so excited that I was working through most of the night as well. By the end of the following week, the structure was solved. Extraordinary as it seemed to me, the Harvard proposal was correct. The rings were parallel, with the iron atom sandwiched between them at a crystallographic centre of symmetry. There was no doubt about it. That was the marvelous thing about crystal structure analysis. When it worked, the result had a satisfying definiteness about it. Even though this aura of definiteness could sometimes be misleading! The crystalline structure of ferrocene occupied me, on and off, for more than thirty years. Later, the apparently staggered orientation of the cyclopentadienyl rings was revealed to be an artifact resulting from crystal disorder.[26]Ferrocene turned out to be trimorphic — at least — and the ring orientation in the low-temperature stable polymorph is eclipsed not staggered.[27]
Back to 1952: there was still the question of how to account for the new kind of bonding in this extraordinary molecular structure. How can the iron atom simultaneously make ten Fe—C bonds? How could the tenfold symmetry be reconciled with the well-known tendency of Fe2+ to form 4- or 6-coordinated complexes? Faced with this challenge, within a few days Leslie developed an explanation based on orbital symmetry properties, on the relationships between the symmetry properties of the d-orbitals of the metal atom and he π-molecular orbitals of the cyclopentadienyl rings. This was new terrain. This new type of molecule required a new type of description of its bonding, and Leslie's model, formal and over-simplified as it was, expressed the essence of this. When it was first explained to me I did not understand a word, but by the end of the week I had picked up enough of the group theoretical background of this new language to construct simple statements on my own. In particular, I could see that the model was a generalization of the standard molecular orbital (MO) model of benzene and other aromatic systems. So we wrote a paper, covering both the structure determination and the new theoretical model, and sent it off to Nature on July 2nd, less than a month after we had the crystals, under the provocative title, "Bis-cyclopentadienyl Iron: a Molecular Sandwich".[28] That was the first time, I believe, that this gastronomic epithet had been used in the title of a chemical publication. The name certainly stuck.
I have the obligation to try to make amends for having then deprived my co-author of credit for what, at that time, would have turned out to be an important theoretical prediction. Since the same symmetry arguments as we applied to ferrocene could be applied mutatis mutandis to the then still unknown and scarcely imagined molecules dibenzene chromium and dicyclobutadiene nickel, Orgel wanted to include in our paper his prediction that these molecules would turn out to be stable species. I argued that it would be a pity to spoil a good, solid paper by what could be regarded as risky speculation and managed to persuade him to omit the additional paragraphs. As Leslie later ruefully remarked, one characteristic of our collaboration was that we sometimes succeeded in shooting down each other's best ideas.
There is a nice if-then-what? postscript to this story.[29] In September 1951 Pauson and I happened to meet at the IUPAC meeting in New York. He had with him a crystalline sample of dicyclopentadienyl iron, which he intended to hand over to my former research director, Professor J. Monteath Robertson, in the hope that he would be able to establish the correct molecular structure. Robertson, on his way to Cornell University to give the Baker Lectures, was one of the plenary lecturers at the IUPAC meeting. Pauson and I happened to meet at Robertson's lecture. As we had both been students at Glasgow University, we knew one another and sat together. Pauson, according to a letter he wrote to me in 1990, nearly forty years later, considered whether he might entrust his precious crystals to me rather than to Robertson, as he had first intended. However, with the crystals "burning a hole in his pocket", he decided to stick to his original plan. He entrusted the crystals to Robertson. After his arrival at Cornell, Robertson gave the material to Lynn Professor J. Lynn Hoard, and Hoard then passed them on to a student, who was not successful in solving the structure. By the following summer, when I became involved in the ferrocene work, I had forgotten about our meeting in New York and was in any case completely unaware of Pauson's predicament. Now we come to the "if" possibility. If Pauson had given the crystals to me at the New York meeting, history would have taken a different course. Instead of wasting my time after my return to Oxford with my direct methods approach to the calciferol problem, I could have started work on Pauson's crystals almost immediately. It is quite likely that the sandwich structure would then have established within a few weeks, in time for Pauson to revise the paper he had sent to Nature with the wrong structure and almost certainly well before March 1952, in which case Woodward and Wilkinson would never had had the opportunity to publish their audacious structure proposal. I wonder what I would have thought when the unprecedented sandwich structure began to take semblance in the electron density maps.[30]
Besides our work on ferrοcene, Orgel and I wrote at that time a paper about hydrogen bonds. It was then generally considered that O-H···O hydrogen bonds were unsymmetrical, with the hydrogen atom closer to one oxygen atom than to the other. We proposed that the acid maleate anion should have an unusually strong, symmetrical hydrogen bond, and backed this up with spectroscopic observations on crystalline potassium hydrogen maleate.[31] Our proposal was subsequently confirmed by neutron diffraction studies.[32] This may be the first example of what has come to be known much later as a low barrier hydrogen bond (LBHB).
In September 1952, on the invitation of my old Caltech friend Edgar Heilbronner, now returned to the ETH in Zurich, I visited there for the first time and lectured on my work on cyclobutane and on ferrocene. I had no idea then that I would spend the greater part of my life there.
Around that time, we used to discuss more biological themes with the budding molecular biologist Sydney Brenner, who arrived in Oxford in the autumn of 1952 to work in Hinshelwood's laboratory on bacteriophage. This was an area about which I had then a smattering of knowledge through my earlier contacts with the Delbrück group at Caltech. So Sydney talked to me, and I talked to Leslie Orgel, and it was not long before we were talking together — sometimes all three at once — about possible roles of proteins and nucleic acids, and the growing evidence for the involvement of DNA as the carrier of genetic information. Of course, our education in these areas was sadly incomplete and fragmentary, but what we lacked in erudition we made up for in fantasy. We argued about whatever we happened to have learned, especially about the possible meaning of the latest results from the latest journals, or rather, as I recall, Leslie and Sydney argued while I acted as a kind of umpire. It was tremendous fun.
At the same time, my connection with the crystallographers in Cambridge brought me every few months into exciting though inconclusive discussions over pub lunches with Francis Crick. Hence I was aware that he and Watson, whom I had known from Caltech, were trying to deduce the structure of DNA by model building but without any reliable diffraction data to test their models. As I recall, their mood oscillated wildly between enthusiastic optimism and downcast pessimism. I did not give them much chance. In late 1952 I advised Watson to abandon the project and get down to some more promising project. Naturally, I reported on this work to my Oxford discussion partners. At any rate, in early April 1953, when Francis telephoned to ask me to come to look at their marvelous new DNA model, all three of us traveled together to Cambridge, together with Dorothy Hodgkin and her young assistant Beryl Ougton. We knew enough about the problem to recognize almost immediately that the proposed DNA structure must be correct in its essential features. Did we realize that we were present at the dawn of a new age? Did we feel: "At this place and on this day a new epoch in the history of the world begins, and we shall be able to say that we were present at its beginnings"? (These are Goethe's words, written on September 20, 1792, the occasion being the defeat of the Prussian army by the ragged French militia at the battle of Valmy.)
In August 1953 I married Barbara Steuer and returned almost immediately to Pasadena for the second stay, as described above.
INTERIM (1954-1957)
The three years between the end of my second stay in Pasadena (August 1954) and my move to Zurich (October 1957) were approximately equally divided between the U.S. National Institutes of Health in Bethesda, Maryland, and the Davy Faraday Laboratory at the Royal Institution in London. At Caltech I had worked for a time with Alex Rich, who wished to learn something about diffraction methods for studying molecular structure. For instruction purposes, Pauling had placed him under my supervision, carefully adding that I would be responsible for any damage caused to X-ray equipment. One of Alex's first tasks was to visually estimate intensities of reflections on my X-ray diffraction photographs from ferrocene crystals, a task that he passed on to his wife Jane, who thus earned our indebtedness.[33] Alex was offered a position to develop structural research under Seymour Kety at the National Institute of Mental Health and asked me to help setting up the new laboratory. My wife and I were attracted by the idea of spending some time on the East Coast and also by the offer of a post of Visiting Scientist at a salary more than double what I was earning as Research Fellow at Caltech, so I accepted Alex's proposal.
NIH was a completely new world for me. In the first place, in contrast to what I had previously experienced, there was no shortage of funds to buy the latest scientific equipment. And moreover, since most of the scientists there lacked any deep knowledge of physical chemistry, I found myself suddenly transformed into an expert, relatively speaking, on this branch of science. Accordingly, for example, I was called on to give advice on problems in infrared, visible and ultraviolet spectroscopy, subjects in which I possessed only a smattering of knowledge. After a month or two, Alex left for more exciting collaboration in Cambridge with Francis Crick on the structure of collagen and I was left to my own devices. I learned that sodium dithionite Na2S2O4 was much used as a reducing agent in biochemistry but also that no one seemed to know the structure of the compound or why it was a reducing agent. Since it was a solid, I decided to determine its crystal structure. This information indeed provided the answer to the problem. The anion turned out to be a dimer of SO2- with a remarkably long S-S bond. In solution, deprived of the surrounding shell of sodium ions, the dimer would clearly dissociate into SO2-, which would then donate its extra electron to any suitable acceptor.[34] In support of this, sodium dithionite is diamagnetic but its solution shows an ESR signal that gradually decays.[35]
Most X-ray crystallographers have never seen a burn caused by exposure to X-rays. I have. During the experimental work on sodium dithionite, I committed a stupid sin of carelessness in the laboratory. This was just after the birth of our daughter Marguerite. Perhaps my main attention was directed towards her. While adjusting a crystal on its mount, I noticed that the X-ray window was open and realized that my fingers must have been exposed to the invisible radiation. Naturally, I closed the window immediately but could not help asking myself how much damage had been done. An X-ray burn is different from an ordinary burn; as there is no heat there is no charring of the skin or flesh. The damage is deeper down and takes some time before there is any outward expression. After a few days, the thumb and forefinger of my left hand began to itch. Erythema soon developed into swelling and formation of blisters. The medical staff of the NIH Radiation Safety Office were outspokenly pessimistic about the outcome of my accident. They talked about possible amputation of the injured finger and thumb because of radiation damage to the underlying bone. It gradually became clear to me that the experience of the medical staff was limited to burns caused by high-energy X-rays, the kind used in medical X-ray equipment. The doctors reasoned that for an X-ray exposure to produce such extensive skin damage as in my case, the damage to the bone must be considerable. I knew this reasoning was wrong. The radiation produced by a copper X-ray tube (the kind I was using) would be almost entirely absorbed by the upper skin layers. For copper radiation, with a linear absorption coefficient of about 12 cm-1 for water and organic matter, only a tiny fraction of the incident intensity would penetrate to any depth and be absorbed by the bone tissue. Therefore the doctors, in judging from the skin damage, were vastly overestimating the damage to the underlying bone, protected by, say, half a centimeter of tissue and fluid. I insisted that I wished to keep my damaged fingers. But as the weeks passed, and the open sores that developed became infected, I did become worried. The healing process was slow, but all that was left to remind me of my stupid error was scar tissue on the fingertip. Later, I used to show this to beginning students as a warning.
At NIH my wife and I were much occupied with the question: should we stay in the U.S. or return to Europe? There was much to be said on both sides. Since our married life had been spent entirely in America, far from our families, we felt we needed some experience of life together in Europe. I wrote to Dorothy Hodgkin, asking if she knew of any suitable openings. She informed me that Sir Lawrence Bragg, recently retired from his professorship at the Cavendish Laboratory in Cambridge, was building a new research group to carry out crystallographic research at the Royal Institution in London. I applied and soon received a warm and enthusiastic answer from Bragg. In a letter dated August 12, 1954, he described the preliminary results obtained by Perutz on haemoglobin and by Kendrew on myoglobin; the final paragraph read: "There is every chance that with a concerted drive on the problem, light will suddenly break and some key to the general structure of protein emerge. The more good people we get to work together here the better, and if you would like to be one of them, I will do all I can to make it possible." I replied that although I was very interested in the possibility of working at the Royal Institution, I was not enthusiastic about the prospect of concentrating exclusively on protein research. "Had I complete freedom to choose a line of research for myself, I do not think I would concentrate on protein analysis. I might choose to apply modern X-ray techniques to some of the problems of inorganic structural chemistry which survived the attacks by yourself and others in the early days of crystallography. Especially in the complex coordination compounds of the transition metals there are many problems left. Another study interesting to me would be that of the specificity of molecular compound and complex formation, where apparently very weak forces can achieve a high degree of specificity. Both of these fields would, of course, tie in with the study of proteins although in an indirect fashion. The first would be a good starting point for finding out something about the important prosthetic groups containing transition metals. The second might tie in with a study of the specificity of enzyme systems. But perhaps this is building castles in the air, and I only mention it to emphasise the fact that my present interests are in regions far from direct protein structure analysis."
Today I am not displeased with this statement of my intentions, but I had serious misgivings about it then, once my letter had been posted. While I did not wish to tell outright lies to Bragg, surely a little prevarication would have been preferable to this blunt statement. As the months went past without any word from London, the prospect of joining Bragg's group at the Royal Institution became more and more remote. I began to look into other possibilities.
In April 1955, Bragg wrote to apologize for his delay in replying to my letter and offered me a five-year appointment as Senior Research Fellow at the Davy-Faraday Research Laboratory. He wrote:"I cannot see my way clearly in all directions yet. I am not clear of the financial tangle and many plans are still uncertain, but if you would like to join in the venture and help with the research side, I will do all I can to give you a good time. Once again, I am very sorry I did not write to you at once. I could not have been more definite about plans at that stage but I should have acknowledged your letter."
With all my misgivings about the tone of my letter, here was Bragg apologizing to me! I agreed to join and suggested that I might initiate X-ray work on haem itself or some other simple porphyrin derivative (sans protein!), a proposal that found immediate favor with Bragg, although nothing ever came of it because of lack of suitable crystals. We moved to London in January, 1956.
I knew from the start that I was going to enjoy my work at the Royal Institution. With its superb position and noble I in Albemarle Street, just off Piccadilly, round the corner from Burlington House, the home of the Royal Academy, the Royal Society and many other learned societies, it was obviously a most attractive place to work. Its well-stocked and renovated library and reading room and its lecture theatre, surely one of the most beautiful anywhere, added to its attraction. And most of all, I felt the historic aura of its connection with the past century and a half of science. At the back of the building was the Davy-Faraday Research Laboratory, situated on several floors to which one gained access by an old, dangerous-looking, rope-drawn lift. The basement contained the X-ray facilities, including impressive high-voltage equipment protected by a metal wire trellis. I forget how many kilovolts and kilowatts could be generated but I have the impression that the X-ray intensities produced were at that time the highest available anywhere. My office was on the third floor. For the first few weeks I had it to myself, but then I was joined by David Phillips, just arrived from Canada, who was to become famous with his X-ray crystallographic analysis of lysozyme. We became life long friends. Other members of Bragg's team were Uli Arndt (involved in the development of the linear diffractometer and computer controlled diffractometers), Tony North, David Green, and Helen Scoloudi, formerly with Bernal and Dorothy Hodgkin, and all presided over by the benign Sir Lawrence.
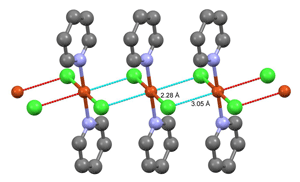
Through conversation with Ronald Nyholm, recently appointed to the chair of inorganic chemistry at University College, London, I decided to study the crystal structures of cobalt dipyridine dichloride, CoPy2Cl2 and its copper analogue CuPy2Cl2. There were two known forms of the cobalt compound, one violet coloured, the other blue. The blue form was known to contain discrete molecules with tetrahedral bonds at the cobalt atom, while the violet form was believed to contain polymeric chains with octahedral bonds at the cobalt atom, each chloride ion linked to two cobalts, each cobalt to four equidistant chloride ions and to the pyridines. Indeed, this turned out to be the case. The copper compound was found to have a very similar structure, except that the four chloride ions were not equidistant from the metal atom; instead, there were two short Cu-Cl bonds and two long ones.[36] The structure of CuPy2Cl2 showing two different Cu—Cl distances. (Drawn by Jeff Deschamps from CSD PYRCUC01)
This result led to another collaboration with Leslie Orgel, who, a couple of years earlier, had suggested that such distortions of octahedral complexes could be interpreted in terms of crystal-field theory as structural expressions of the Jahn-Teller effect. The differences between the octahedral coordination in the cobalt compound and the distorted octahedral coordination in the copper compound seemed a perfect illustration of this, and I soon found that similar differences between other pairs of structurally related compounds occurred according to a quite regular pattern.[37] We also saw that crystal field theory could be applied to minerals with the spinel structure. Spinel is a mineral with composition MgAl2O4, built from a cubic close-packed arrangement of oxygen atoms with the Mg2+ ions at tetrahedral cavity sites and the Al3+ ions at octahedral ones. There are many other AB2O4 minerals with essentially the same structure, with the A2+ ions in tetrahedral sites and B3+ ions in octahedral ones. However, in "inverted" spinels the tetrahedral sites are occupied by B3+ ions, with the A2+ ions and the remaining B3+ ones distributed at random over octahedral sites. We found we could explain all the known experimental evidence on the metal ion distributions in the normal and inverted spinels. Moreover, the existence of tetragonally deformed spinels could also be explained by our theory in terms of the Jahn-Teller distortions expected to occur when certain metal ions were present.[38] When I told Bragg about these results he was delighted. He was just then working on a new edition of his classic "The Crystal Structures of Minerals" and could now include an explanation of the problem of the inverted and tetragonally distorted spinels.[39] Probably this helped to reconcile him to my reluctance to work on crystal structure analysis of proteins.
In London I acquired my first two doctoral students. One was Peter Pauling, Linus's son, who had decided to work for his doctorate in Britain, and the other was David Brown, who later became a well-known inorganic crystallographer in Canada. They were officially enrolled as graduate students of the University of London, but because of the delicate and intricate relationship between the University and the Royal Institution, they were nominally under the direction of the professor of Inorganic Chemistry, Ronald Nyholm, although in fact they worked with me. Peter studied the polymorphism of anhydrous cobalt sulfate, three forms with very similar structures but differing in the site symmetries of the tetrahedral sulfate groups.[40] David worked on the structure of the crystalline cuprous chloride-azomethane complex[41] and of diazoaminobenzene copper (I), which turned out to contain a remarkably short Cu···Cu distance of 2.45 Å.[42]
In December 1956 Edgar Heilbronner telephoned out of the blue to ask if I could come to Zurich to talk to Professor Leopold Ruzicka about the possibility of my starting a crystal structure analysis group at the Swiss Federal Institute of Technology (ETH Zurich). Ruzicka was due to retire the following October from his position as Professor of Organic Chemistry. Impressed by Dorothy Hodgkin's success in deciphering the structure of vitamin B12, he saw that a strong organic chemistry team would be incomplete without this new method. Ruzicka offered me a post as associate professor (Ausserordentlicher Professor) with a start-up grant of about 100,000 Swiss Francs for equipment. He explained that he needed a quick reply. He wanted to fill the post before his retirement. If I were unwilling or unable to come, he would look for someone else. He gave me fourteen days to decide. When I returned to foggy London just before Christmas, my wife and I weighed the pros and cons of London and Zurich, England and Switzerland. There was also the problem of my post at the Royal Institution. I had promised Bragg to stay for five years and now I was thinking of leaving after less than a year had passed. Bragg advised me to go, for, as he said, I might never get such an opportunity again. A few days before the end of the year I sent a telegram to Ruzicka to accept the offer. Nine months later, on October 1st, 1957, I began my new career at the ETH.
Jack Dunitz in his office (ca. 2012).
Jenny P. Glusker, Jack D. Dunitz, Carolyn P. Brock, at the celebration of the 40th anniversary of the CCDC (2005). Jack and Jenny were both very important to the success of that organization. (Courtesy of Carolyn P. Brock)
Editor's Note
This essay was sent to ACA RefleXions by Jack Dunitz in 2012. Photographs and drawings to illustrate the text have been added. The text is similar to the essay posted on the Oregon State University website http://scarc.library.oregonstate.edu/coll/dunitz/primavera/page1.html and to the recent article in Helvetica Chimica Acta, DOI: 10.1002/hlca.201300038. Also see the appreciation of Dunitz in the same issue by W. Bernd Schweizer and Ryan Gilmour, DOI: 10.1002/hlca.201300092.
References 3 Nordman, C. E.; Lipscomb, W. N. J. Chem. Phys. 1951, 19, 1422.
4 Dunitz, J. D. Chem. Eur. J. 1998, 4, 745.
6 Bernal, J. D.; Crowfoot, D.; Fankuchen, I. Trans. Roy. Soc. (London) 1940, 239, 135; Crowfoot, D. Vitamins and Hormones 1944, 2. 409.
14 Pauling, L.; Brockway, L. O. J. Amer. Chem. Soc. 1937, 59, 1223; Donohue, J.; Humphrey, H.; Schomaker, V. ibid. 1945, 67, 332.
18 Phage and the Origins of Molecular Biology (Ed. Cairns, J; Stent, G. S.; Watson, J. D.), Cold Spring Harbor Laboratory of Quantitative Biology, 1966.
20 Pauling, L; Corey, R. B.; Branson, H. R. Proc. Nat. Acad. Sci. U. S. 1951, 37, 205; Pauling, L; Corey, R. B. ibid. 1951, 37, 235, 241, 251, 256, 261, 272, 282, 729.
32 Darlow, S. F.; Cochran, W. Acta Cryst. 1961, 14, 1250; Fillaux, F.; Leygue, N.; Tomkinson, J.; Cousson, A.; Paulus, W. Chem. Phys. 1999, 244, 387.
35 Hodgson, W.G.; Neaves, A.' Parker, C. A.; Nature (London) 1956, 178, 489; Rinker, R. G.; Gordon, T. O.; Mason, D. M.; Corcoran, W. H. J. Phys. Chem. 1959, 63, 302.
|